![]()
Mitglied bei

Eine weitere Suchmöglichkeit besteht darin, z.B. bei www.google.de das Suchwort einzugeben und dann nach einem Leerzeichen den Zusatz site:www.cfs-aktuell.de
Sie erhalten dann alle Seiten auf cfs-aktuell.de, auf denen der gesuchte Begriff vorkommt.
Artikel des Monats August 2012 Teil 4
Die "Wiederentdeckung" des XMRV/PMRV/MLV
Deutsche Forscher: Es war keine Laborkontamination...
|
Bitte beachten Sie: 2012 hat sich herausgestellt, dass dieses XMRV keine Humaninfektion, sondern eine im Labor entstandene Chimäre war. Näheres unter Artikel des Monats Dezember 2012 - 1 auf dieser Website! |
Die Katze, die die Mäuse fing
Aus: http://me-advocacy.com/Cat_that_caught_the_mice.html, Übersetzung Regina Clos
|
Anm.d.Ü.: Wenn Sie diesen hochtechnischen und für den Laien reichlich unverständlichen Artikel zu einer deutschen Studie über die angeblichen Vorläuferviren der angeblichen Laborkontamination XMRV nicht ganz lesen wollen, dann springen Sie am besten gleich zu den Schlussfolgerungen unten. Und lesen Sie auch den neuesten Blog von Jamie Deckoff-Jones, die hier ebenfalls zu dem Artikel der deutschen Forscher Mayer et al. von der Universität des Saarlandes Stellung nimmt und aus ihr den Schluss zieht, dass der Großteil der bisherigen Arbeiten, die Negativstudien produziert haben, "eine bedeutungslose Verschwendung von Zeit, Energie und Geld" gewesen sei. Und noch ein paar wichtige Informationen, die ich gerade in einem Forum las, in dem fachkundige Insider sich austauschen: Judy Mikovits, die 2009 die bahnbrechende Studie über den Zusammenhang von ME/CFS mit Mäuseleukämievirus-verwandten Retroviren, damals XMRV oder PMRV genannt, entdeckt hatte, wird immer noch daran gehindert, sich zu verteidigen gegenüber all den Negativstudien und den Angriffen auf ihre Forschung und die anderer, die ebenfalls diese Retroviren als infektiöse humane Retroviren identifiziert haben und dies weiterhin tun. Und zwar durch eine Zivilklage ihres früheren Arbeitgebers, dem Whittemore Peterson Institute und weil die Polizei es weiterhin ablehnt, ihr die Notebooks auszuhändigen, auf denen ihre Forschungsarbeiten dokumentiert sind. Wenn Judy Mikovits darüber sprechen würde, was sie weiß und was ihr und anderen Forschern passiert (ist), dann droht ihr Gefängnis wegen Missachtung des Gerichts. Soviel zur Freiheit der Wissenschaft in den USA. Mit ihren Notebooks könnte sie die Existenz der Gammaretroviren beweisen, die mit ME/CFS in Zusammenhang stehen. Diese Retroviren haben und hatten nie etwas zu tun mit dem "XMRV" aus der Zelllinie VP62, die ein künstliches Gebilde ist und niemals bei einem Menschen gefunden wurde - das aber Grundlage und Gegenstand all der Negativstudien war, in denen es natürlich deshalb nie gefunden werden konnte. Hinzu kommt, dass deren Testmethoden größtenteils ungeeignet waren, überhaupt solcherart Retroviren zu finden. Ein paar Halb- und Teilwahrheiten finden sich in diesem Artikel: http://www.thedailybeast.com/articles/2012/07/23/how-research-into-chronic-fatigue-syndrome-turned-into-an-ugly-fight.html Ob sich mit dieser Studie von Mayer et al. (siehe unten) und mit den mutigen Aussagen von Denise O'Keefe über eine kürzlich erschienene italienische Studie * irgendwann der Schleier vor der wirklichen Bedeutung dieser humanen Gammaretroviren lüften wird? Wir dürfen gespannt sein. Die Lipkin-Studie, auf die die ME/CFS-Gemeinde so mit Spannung wartet, ist noch immer nicht veröffentlicht. Und, Insider vermuten, dass sie aus verschiedenen Gründen wie etwa ungeeigneter Probensammlung und -verarbeitung auch "negativ" ausfallen wird. Sobald hier etwas Neues bekannt wird, erfahren Sie es auf dieser Website. R.C. * In dieser Studie hat man bei zwei von 12 ME/CFS-Patienten Sequenzen aus der MLV-GAG Region gefunden und sorgfältig jede Laborkontamination ausgeschlossen... |
Dr. Coffin und Dr. Pathak haben ihre Meinung über die Herkunft des XMRV zuerst bei der CROI Konferenz 2011 (1) geäußert. Später gaben sie ein Video (2) heraus, in dem erzählt wurde, wie sie die Geburt des Virus auf eine Zelllinie mit dem Namen 22Rv1 zurückverfolgt hatten und wie sie auf wundersame Weise zwei parentale (elterliche) Viren für das XMRV (PreXMRV-1 und PreXMRV-2) identifiziert haben. Es war eine fantastische Geschichte mit allen Merkmalen einer wundersamen Entdeckung, die nur die scharfsinnigsten und konzentriertesten Geister entdecken konnten – zumindest war das der Eindruck, den viele durch die Publizität des Artikels von Paprotka et al. 2011 (3) gewinnen mussten.
Eine nähere Untersuchung der Experimente, die in dem Artikel ‘Recombinant origin of the retrovirus XMRV’ (3) dargestellt werden, konnte diese Behauptungen jedoch nicht stützen, und die Sequenz, die in den GenBank eingetragen wurde und von der gesagt wurde, sie stamme aus der Zelllinie 22Rv1und die als „Konsens“-XMRV-Sequenz bezeichnet wurde, enthielt kein env-Gen. Man hat dabei auch entdeckt, dass XMRV nicht das Virus war, das in der Humanpopulation von verschiedenen Wissenschaftlern entdeckt worden war. Diese Viren waren tatsächlich XMRV-ähnlich.
Anfang dieser Woche haben Wissenschaftler aus Deutschland (Mayer et al. 2012 (4) ) eine Studie veröffentlicht, in der nach einem der Viren gesucht wurde, von denen Paprotka et al. behauptet hatten, sie seien das Vorläufervirus des XMRV gewesen. Die Forschungsergebnisse dieser Wissenschaftler jedoch widersprechen direkt dem, was Paprotka et al. (3) und Cingoz et al. 2011 (5) als Ursprung dieses Vorläufervirus und den humanen XMRV-ähnlichen Sequenzen erklärt hatten.
Eine kurze Zusammenfassung der Studien von Paprotka et al. (3) und Cingoz et al. (5) wird am Ende dieses Artikels bereitgestellt. Die wichtigsten Punkte werden hier so als Aufzählung wiedergegeben, wie sie die deutsche Studie betreffen.
-
Es wurden in keiner Studie wilde Mäuse untersucht
-
In beiden Studien wurde nur PCR (Polymerasekettenreaktion) eingesetzt
-
45 Labormäuse und 44 von wilden Mäusen abstammende Labormäuse wurden von Paprotka et al. negativ auf XMRV getestet.
-
15 Labormäuse wurden von Paprotka et al. auf PreXMRV-1 und PreXMRV-2 getestet (darunter keine von wilden Mäusen abstammenden Labormäuse). Mehrere Stämme waren positiv.
-
48 Laborstämme und 46 von wilden Mäusen abstammende Laborstämme wurden von Cingoz et al. auf PreXMRV-1 und -2 getestet. Mehrere Stämme waren positiv.
-
Die 22Rv1 Zelllinie wurde unter Verwendung von Nacktmäusen geschaffen.
-
Cingoz et al. fanden heraus, dass die sogenannten PreXMRV-1 und -2 Viren bei behaarten Mäusen, jedoch nicht bei Nacktmäusen integriert waren.
-
Paprotka et al. behaupten, dass PreXMRV-1 und PreXMRV-2 endogene Mäuseviren seien.
|
ERGEBNISSE DER DEUTSCHEN STUDIE Virus Res. 2012 Jul 4. [Epub ahead of print] Comparing PreXMRV-2 gag sequence diversity in laboratory and wild mice using deep sequencing. (Vergleich der Diversität der PreXMRV-2-gag-Sequenzen bei Labor- und wilden Mäusen mit Hilfe von Deep Sequencing) Mayer J, Mazzoni CJ, Greenwood AD. Quelle: Abteilung für Humangenetik , Zentrum für Human- und Molekularbiologie Medizinische Fakultät der Universität des Saarlandes, 66421 Homburg, Deutschland Zusammenfassung Es wurde kürzlich berichtet, dass das Xenotrope murine Lekämievirus-verwandte Virus (XMRV) einer Rekombination im Labor entstammt. Jedoch wurden Sequenzen mit den Charakteristika der 5‘-Hälfte des XMRV (als PreXMRV-2 bezeichnet) im Genom mehrerer Labormäuse und Zelllinien identifiziert wurde, was darauf schließen lässt, dass Teile des XMRV-Genoms als natürlich vorkommende Retroviren bei Mäusen existieren. Wir vergleichen hier die Diversität der PreXMRV-2-gag-Sequenzen in Mäusen mit den berichteten XMRV-ähnlichen Sequenzen, indem wir die genomische DNA einer Reihe von Stämmen von wilden Mäusen und von verbreiteten Inzuchtstämmen von Labormäusen mit Hilfe von Hochdurchsatz-Amplicon-Sequenzierung testen. Sequenzen mit Merkmalen, die typisch sind für die zuvor berichteten PreXMRV-2-Sequenzen, darunter einer 24-nt-Deletion, wurden bei verschiedenen wilden Mäusen und bei Inzucht-Mäusestämmen innerhalb von hochgradig nicht XMRV-ähnlichen Sequenzen wiederholt identifiziert. Eine Sanger-Sequenzierung der Klone von Amplikonen scheiterte jedoch dabei, solche Sequenzen effektiv wieder aufzufinden. Eine phylogenetische Analyse lässt darauf schließen, dass PreXMRV-2-gag-Sequenzen von Mäusen, Zelllinien und Patientenproben zum gleichen evolutionär jungen Stamm gehören und dass diese Sequenzen unterschiedlich sind und weit verbreitet bei Mus musculus domesticus (Hausmaus) und Labormäusen, die von dieser Spezies abstammen. Es konnten keine Belege für PreXMRV-2-ähnlichen gag-Sequenzen außerhalb der Mus Musculus-Abstammungslinie gewonnen werden. Die Ergebnisse lassen darauf schließen, dass eine genaue Bestimmung des Vorhandenseins, des Fehlens und der Beziehungen von spezifischen murinen retroviralen Stämmen sehr stark von einer Analyse mit Hilfe von Deep Sequencing [Gen-Sequenzierung der nächsten Generation, Hochdurchsatz-Sequenzierung, d.Ü.] profitieren können. |
Mayer et al. verglichen die Diversität (Vielfältigkeit, Unterschiedlichkeit) der PreXMRV-2 und XMRV-ähnlichen Sequenzen bei Mäusen – sowohl bei durch Inzucht erzeugten als auch bei wilden Mäusestämmen und auch bei einer Reihe anderer Nagetiere. Sie setzten dabei sowohl Tests mit geringem Durchsatz (PCR) als auch Tests mit hohem Durchsatz (Next Generation Sequencing – NGS) ein. Im Vergleich zur PCR produziert die Next Generation Sequenzierung viele Millionen mehr Sequenzen eines Virus, wodurch sie ein viel besserer Ansatz zur Entdeckung einer geringgradigen Infektion mit einem spezifischen Virus ist als die veralteten PCR-Techniken. Überraschenderweise wurde bislang bei keiner Studie diese Sequenzierung der nächsten Generation eingesetzt, um die Diversität der MLVs innerhalb der Mäuse oder bei verschiedenen einzelnen Mäusen zu kartieren.
Nach Aussage der Autoren wurde das PreXMRV-2-Virus gewählt, da die Experimente rund um die 24-Nukleotid-Deletion in der gag-Region des Virus herum konstruiert werden konnten. Für diejenigen, die es noch nicht wissen – PreXMRV-2 ist ein polytropes Virus, dass in der gag-Region und in Teilen der pol-Regionen dem XMRV zu 99,9% ähnlich ist, das auch eine 24-Nukleotid-Deletion in seiner gag-Region hat. Es ist auch wichtig, sich zu vergegenwärtigen, dass die Zahl von 67% positiv getesteten ME/CFS-Patienten in der Studie von Lombardi et al. Von 2009 (6) mit einem Assay gewonnen wurde, das auf die gag-Region des VP35 abzielt, einem Virus mit einer hohen Ähnlichkeit (99,8%) in der gag-Region zu PreXMRV-2. Von den zwei Primer-Paaren, die in dieser Studie verwendet wurden, ist das zweite längere Set darauf angelegt, die gag-Region des humanen Retrovirus VP35 zu entdecken.
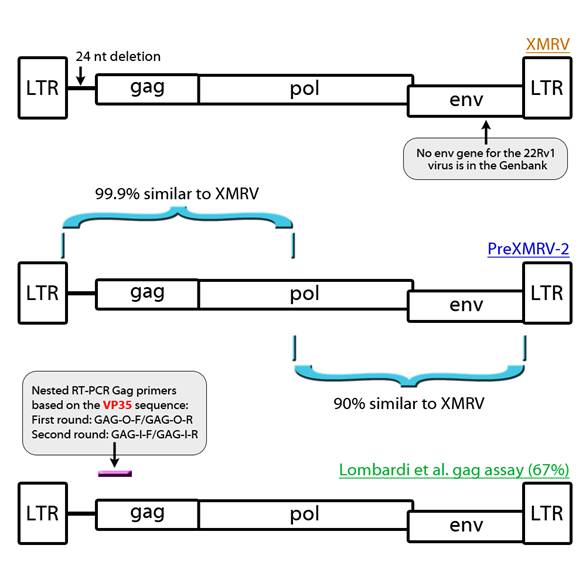
Mayer et al. entdeckten, dass da, wo die Verfahren der Gen-Sequenzierung der nächsten Generation MLV-Viren entdecken konnten, die PCR, die in den Studien von Paprotka und Cingoz verwendet wurde, im Allgemeinen nichts entdeckt. Es wurden hierfür zwei Gründe vorgeschlagen: die geringe Kopienzahl der Viren und die teilweise Deletion (Löschung) in der gag-Region der Viren, die die Spezifität der verwendeten Assays beeinträchtigt.
Die Autoren stellten auch heraus, dass Cingoz et al. behauptet hatten, XMRV-ähnliche Sequenzen in verschiedenen Mäusen identifiziert zu haben, sie aber tatsächlich nicht versucht hatten, die Produkte, die durch die PCR amplifiziert (vermehrt) worden waren, zu sequenzieren, was die Identifikation der Viren zu einer reinen Annahme verkommen lässt.
Ebenfalls im Widerspruch zu den Befunden von Paprotka und Cingoz, die PCR eingesetzt hatten, fanden Mayer et al. auch heraus, dass – wenn man eine Sequenzierung der nächsten Generation durchführt - das PreXMRV-2 eine Sequenzdiversität bei wilden und bei Inzuchtmäusen hat. Das weist darauf hin, dass PreXMRV-2 kein endogenes Mäusevirus sein kann.
“die vom Menschen abgeleiteten, von der Zelllinie abgeleiteten und von der Mäuse-DNA abgeleiteten XMRV-ähnlichen Sequenzen zeigten eine schlechte Baumstruktur, eine hohe Diversität in der Zahl der Abstammungslinien und bildeten keine Cluster nach DNA-Quelle…“
Ihre phylogenetische Analyse weist auch darauf hin, dass die meisten Sequenzen, die die 24-Nukleotid-Deletion von Mäusen enthielten und die von Mäusen, der Zelllinie und Humangewebe stammten sowie die Sequenzen ohne Deletion alle von einem unbekannten Stamm abstammten, der eine Mischung von polytropen und xenotropen Sequenzen enthielt. Mehrere Sequenzen waren ähnlich, aber unterschieden sich vom PreXMRV-2-Virus nach der 24-Nukleotid-Deletion und standen den Xenotropen murinen Virus-Sequenzen näher. Ein Hinweis darauf, dass dies rekombinante Sequenzen waren, die ein PreXMRV-2-Virus und ein xenotropes Virus kombinierten. Etwas, das häufig vorkommt bei Viren, die eine hohe Rekombinationsrate haben. Infolge der hohen Ähnlichkeit der Sequenzen glaubt man auch, dass es sich um einen jungen Stamm handelt, was bezogen auf Gamma-Retroviren ein paar Hundert Jahre wären.
“Das PreXMRV-2-Virus hat seinen Ursprung wahrscheinlich in wilden Populationen von Mus domesticus (Hausmaus) und kommt möglicherweise nicht in allen Populationen vor.”
Anhand der getesteten Mäuse fanden sie heraus, dass PreXMRV-2 wahrscheinlich wahrscheinlich von Mus musculus domesticus (der westeuropäischen Hausmaus) abstammt. Dass mehrere Labormäuse und Inzuchtstämme von Labormäusen ebenfalls infiziert waren. Etwas, was man erwarten kann, da die Mehrzahl der Inzucht-Labormäuse von der westeuropäischen Hausmaus abstammt und einige davon das PreXMRV-2-gag mitschleppen.
Die Autoren testeten auch eine Probe von einem Museumsexponat einer wilden Maus (Mus musculus domesticus), die im Jahr 1906 in Ann Arbor Michigan, USA, gefangen worden war. Die PCR-Produkte von dieser Probe ergaben eine Infektion mit einem Virus des Stamms der PreXMRV-2-Familie, aber aufgrund des Alters der Probe und dem dann folgenden Zerfall der DNA konnten sie die Probe nicht sequenzieren. Was sie dazu führte, festzustellen:
“wir können nicht darauf schließen, dass PreXMRV-2-ähnliche Sequenzen in der wilden Hausmaus-Population der USA nicht vorkommen.“
Die Autoren weisen darauf hin, dass die PreXMRV-2-gag-Region möglicherweise weit verbreitet sind bei verschiedenen Unterarten von Mus musculus, die in dieser Studie nicht getestet worden waren.
Sie zogen den Schluss, dass das Virus bei Mäusen nicht mit einer hohen Kopienzahl vorkommt und dass deshalb die Hochdurchsatz-Sequenzierung (NGS – Next Generation Sequencing) generell eingesetzt werden sollte, wenn man versucht, die seltenen Sequenzen dieses Typus zu entdecken. Die NGS wird auch gerne für die Entdeckung von seltenen Varianten des HIV-1 eingesetzt.
Die Autoren schlussfolgerten, dass:
“Das Vorliegen eines eigenständigen Stammes einschließlich XMRV, PreXMRV-2, aus Zelllinien abgeleiteten und aus Mäuse-DNA abgeleiteten Sequenzen lassen darauf schließen, dass die 24-nt-Deletion und die gag-spezifischen Merkmale infolge einer gemeinsamen Herkunft bei allen vorhanden sind und nicht infolge einer unabhängigen Entstehung der Deletion oder anderer Polymorphismen innerhalb dieser Gruppe.“
Und noch einmal: die Hauptschlussfolgerungen der Studie sind:
-
Beim PreXMRV-2 gibt es eine Diversität.
-
Mit Hochdurchsatz-Sequenzierung kann man die MLVs identifizieren, die man mit PCR nicht findet.
-
PreXMRV-2 entstammt wahrscheinlich in Mus musculus domesticus.
Schlussfolgerungen für humane Gamma-Retrovirus-Infektionen
Diese Forschungsarbeit ist der letzte Sargnagel für die Story, dass die ME- und Prostatakrebs-Retroviren eine reine Laborkontamination seien. Die Viren sind nicht beschränkt auf das Labor. Es ist hochwahrscheinlich, dass Rekombinationsereignisse mit PreXMRX-2 in der freien Natur ständig vorkommen, mit anderen polytropen und xenotropen Mäuseleukämieviren (MLVs). Wenn diese Studie etwas zeigt, dann das, dass ohne eine vollständige Sequenzierung der ME-Retroviren und von mehr Prostatakrebs-Retroviren keine Behauptungen über die Varianten und Stämme gemacht werden können, die entdeckt wurden.
Außerdem, wenn MLVs beim Einsatz von PCR nicht immer entdeckt werden können, wie können wir dann erwarten, dass Forscher korrekt bestimmen, ob die gleichen Viren den Menschen infizieren, wenn sie diese mangelhaften Techniken einsetzen? Die Negativ-Studien sind hinfällig.
Es ist vieles noch nicht bekannt über diese Viren, aber wir wissen jetzt, dass die Hochdurchsatz-Sequenzierung eine Grundvoraussetzung für zukünftigen Studien sind, um die Wahrheit aufzudecken. Es ist schon zu viel Geld und Zeit bei der Suche nach Viren verloren gegangen, die einer Entdeckung durch PCR entgehen können.
Literatur
2. NCI. 2011. Cancer Research Now: Investigating XMRV. Youtube.
3. Paprotka et al. 2011. Recombinant Origin of the Retrovirus XMRV. Science. 333, 97-101.
5. Cingoz et al. 2012. Characterization, Mapping and Distribution of the Two XMRV Parental Proviruses. JVI. DOI:10.1128/JVI.06022-11.
PAPROTKA ET AL. 2011 AND CINCOZ ET AL. 2011
Die Behauptung in dem Artikel von Paprotka war einfach. Eine lineare Serie von Zellen, die ursprünglich von einem Prostatakrebspatienten stammten und die durch Mäuse passiert worden waren, sind mit verschiedenen Tests analysiert worden. Es gab Zellen aus den frühen Passagen und aus späteren Passagen durch Mäuse, und eine dritte Gruppe von Zellen, die viel später geschaffen wurde. Sie konnten das XMRV in den früheren Zellen nicht finden, aber in beiden Gruppen der späteren Zellen. Sie haben auch behauptet, dass zwei endogene Viren (PreXMRV-1 und PreXMRV-2) bei Mäusen vorhanden waren, die in ihren Worten „wahrscheinlich eingesetzt worden sind“ bei der Schaffung der 22Rv1-Zelllinie. Drei separate Tests sind durchgeführt worden, um zu dieser Schlussfolgerung zu kommen.
Diese drei Tests wurden jedoch selektiv bei unterschiedlichen Zellen und Mäusen eingesetzt, und jeder hatte eine andere Entdeckungsgrenze. Die beiden ersten Test wurden bei allen Elementen kombiniert eingesetzt, außer bei den späteren Zellen, die durch Mäuse passiert worden waren. Keiner dieser Tests ist jemals als fähig ausgewiesen worden, das XMRV bei einer Kopienzahl von unter 2000 Kopien pro 100 Zellen zu entdecken. Der dritte Test, der der sensitivste von den drei Test war, wurde nur eingesetzt, um die späteren Zellen zu untersuchen, die durch Mäuse passiert worden waren, aber es wurden keine Daten über die Sensitivität dieses Tests vorgelegt (tatsächlich wird dieser Test in dem Artikel nicht einmal benannt), und insbesondere bei diesen Zellen ist niemals bewiesen worden, dass sie vom gleichen Patienten stammen wie die anderen getesteten Zellen.
Bei den untersuchten Mäusen wurden keine wilden Mäusestämme miteingeschlossen, nur verschiedene Stämme von Labormäusen, die von wilden Mäusen abstammten sowie eine Reihe von Labormäusestämmen. Keine dieser Mäuse ist tatsächlich in bei der Schaffung der 22Rv1 Zelllinie verwendet worden.
Zusammengefasst: die Experimente in dieser Studie waren
nicht in der Lage zu bestimmen, dass der Ursprung des Virus in einer einzigen
Zellline zu finden sei. Es konnte auch nicht gezeigt werden, dass die zwei
sogenannten Herkunftsviren überhaupt vorhanden waren, um diese magische
Rekombination zustande kommen zu lassen. Schließlich fand man später heraus,
dass die in die NCBI-Datenbank hochgeladene Sequenz des Virus von der
22Rv1-Zellline kein Envelop-Gen enthielt und von daher nicht behauptet werden
kann, sie sei das XMR-Virus.
Eine spätere Studie von Dr. Coffin (Cingoz et al.
2011) erzählte diese Geschichte von den Herkunftsviren weiter. Er behauptete
hier, dass die Viren in der Genom verschiedener Labormäuse integriert seien,
aber bei diesen Mäusen handelte es sich nicht um Nacktmäuse, die aber für die
Schaffung der 22Rv1-Zelllinie verwendet worden waren.
15. Juli 2012